




carbonylés (dérivés) (suite)
Méthodes de dégradation
Celles-ci peuvent n’être ni oxydantes ni réductrices, ou oxydantes :
a) Dégradations non oxydantes.
1o Les alcalis concentrés coupent les dicétones-β :
R—CO—CH2—CO—R + KOH → R—COOK + R—CO—CH3.
2o Les acides β-cétoniques se décarboxylent par simple chauffage :
R—CO—CH2—CO2H → CO2 + R—CO—CH3.
3o Les acides glycidiques sont dissociés vers 120 °C en CO2 et en aldéhyde :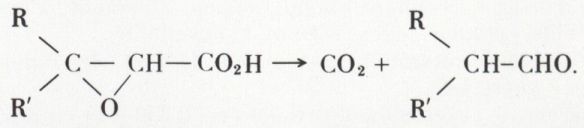
En pratique, ces dégradations constituent la phase finale de préparations synthétiques. L’acétone CH3—CO—CH3 peut être transformée en la dicétone CH3—CO—CH2—CO—CH3 et l’ester acétique en l’ester β-cétonique CH3—CO—CH2—CO2Et. Or, ces deux composés sont facilement alcoylés sélectivement une ou deux fois sur le groupe CH2. Par exemple, sous l’action de l’éthylate de sodium puis de RX, suivie éventuellement d’une nouvelle action de l’éthylate de sodium, puis de R′X, l’acétylaoétone CH3—CO—CH2—CO—CH3 est transformée en 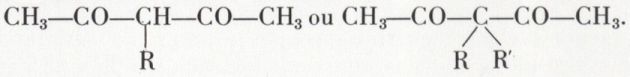
La potasse coupe ces dicétones en CH3—COOK et, respectivement, 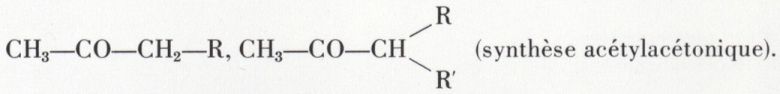
L’ester acétylacétique CH3—CO—CH2—CO2Et se substitue deux fois dans les mêmes conditions et, après hydrolyse, la dégradation, selon 2o, conduit aux mêmes cétones (synthèse acétylacétique).
D’autre part, la cétone R—CO—R′ se condense, en présence d’éthylate de sodium, à l’ester bromacétique :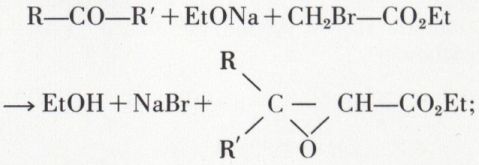
après hydrolyse de l’ester, l’acide glycidique se décompose selon 3o (réaction de Darzens).
b) Dégradations oxydantes.
1o Les glycols-α sont coupés par l’acide périodique ou par le tétracétate de plomb en deux dérivés carbonyles :
R—CHOH—CHOH—R′ + O → R—CHO + R′CHO + H2O.
2o Les alcènes fixent l’ozone :
L’hydrolyse de l’ozonide libère deux dérivés carbonyles :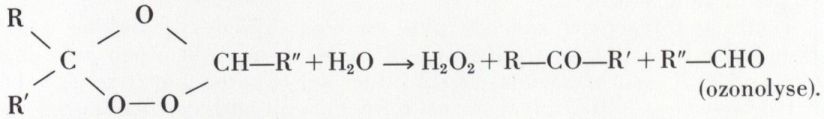
Synthèse éventuellement transpositrice
La déshydratation par l’ion H+ des glycols-α procède par les étapes successives :
Si les quatre radicaux ne sont pas identiques, ce processus fait, a priori, prévoir une double ambiguïté ; toutefois, on a (presque exclusivement)
R—CHOH—CH2OH → H2O + R—CH2—CHO,
CH3—CHOH—CHOH—CH3 → H2O + CH3—CH2—CO—CH3,
C6H5—CHOH—CHOH—C6H5 → (C6H5)2CH—CHO.
Propriétés physiques
Les dérivés carbonyles sont des molécules associées par interaction de dipôle, mais non par liaison hydrogène ; ils sont, en conséquence, plus volatils que les alcools correspondants ; mais CH3—CH2—CH=O est moins volatil que CH3—CH2—CH=CH2 (de même encombrement, mais non associé).
Voici quelques points d’ébullition : HCHO, – 27 °C ; CH3—CHO, 22 °C ; CH3—CH2—CHO, 51 °C ; CH3—CO—CH3, 56 °C ; CH3—CH2—CO—CH3, 78 °C.
En série acyclique, les dérivés carbonylés ont des points de fusion très bas.
Les tout premiers termes sont miscibles à l’eau en toutes proportions ; d’ailleurs, le formaldéhyde s’y combine énergiquement. L’acétone, à la fois hydrosoluble et liposoluble, est un excellent solvant de miscibilité.
Les densités, en série saturée, s’écartent peu de 0,80 : les indices de réfraction ne dépassent guère 1,43.
Le groupe carbonyle absorbe en ultraviolet vers 2 250 Å et en infrarouge entre 1 705 et 1 727 cm–1, nombres qui peuvent s’abaisser notablement si le groupe CO est relié à un radical α-insaturé.
Les premiers aldéhydes ont une odeur suffocante ; l’acroléine CH2=CH—CHO est fortement lacrymogène ; les termes plus lourds sont odorants, et plusieurs d’entre eux constituent des parfums naturels ou artificiels.
Propriétés chimiques
On peut les classer en trois groupes : propriétés strictement fonctionnelles, c’est-à-dire ne faisant pas intervenir les radicaux liés au carbonyle ; propriétés des hydrogènes éventuellement liés aux carbones entourant le carbonyle ; propriétés de l’hydrogène lié au carbonyle (réactions particulières des aldéhydes).
Propriétés fonctionnelles
Le carbonyle est nettement polarisé en
Cette polarisation fait prévoir des additions orientées ; l’électrophile A+ (pratiquement presque toujours H+ ou un ion métallique) s’unit à l’oxygène, et le nucléophile B– au carbone :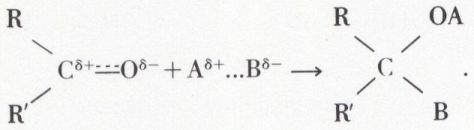
Mais le produit d’addition peut évoluer :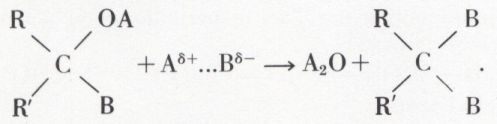
Et si B porte de l’hydrogène (B = B′H),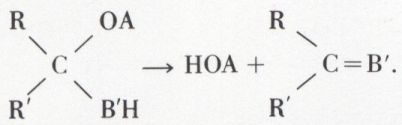
L’addition peut évoluer vers la substitution de O par deux radicaux univalents ou par un radical bivalent.
Toutes ces réactions sont de plus en plus difficiles quand on passe de H—CHO à R—CHO, puis à R—CO—R′ ; elles deviennent à peu près impossibles si R et R′ sont plusieurs fois substitués sur le carbone d’attache ; la cétone (CH3)3C—CO—C(CH3)3 (pivalone) est réfractaire à toutes ces réactions.
Si A = H, elles sont réversibles et à la fois catalysées par l’ion H+, qui transforme R—CO—R′ en l’ion bien plus réactif 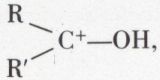 , et par l’ion OH–, qui transforme BH en l’ion B–, plus actif que BH.
, et par l’ion OH–, qui transforme BH en l’ion B–, plus actif que BH.
L’hydrogénation, dont le processus est différent, est un cas particulier.
a) Hydrogénations.
En principe, tout dérivé carbonyle est hydrogénable en alcool :
R—CO—R′ + H2(2H) → R—CHOH—R.
Les milieux alcalins résinifiant les aldéhydes, l’hydrogénation au sodium (en présence d’eau) est réservée aux seules cétones.
Elle est accompagnée d’une faible réduction duplicative :
2 CH3—CO—CH3 + 2 Na + 2 H2O → 2 NaOH + (CH3)2COH—COH(CH3)2 (pinacol).
Cette réduction duplicative devient prépondérante sous l’action du magnésium en milieu aprotonique, suivie d’une hydrolyse :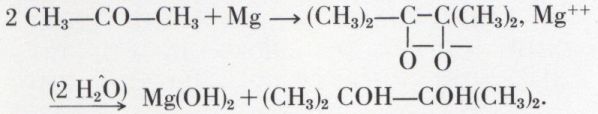
Les acides dilués en présence de zinc sont généralement sans action ; mais les acides concentrés réduisent jusqu’au stade hydrocarbure :
R—CO—R′ + 4 HCl + 2 Zn → 2 ZnCl2 + H2O + R—CH2—R (Clemensen)
[même résultat dans l’emploi de l’hydrazine en milieu alcalin].