Composés renfermant dans leur molécule le groupe carbonyle 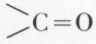 uni soit à deux hydrogènes, soit à un hydrogène et à un radical, soit à deux radicaux, soit aux deux extrémités d’un radical bivalent :
uni soit à deux hydrogènes, soit à un hydrogène et à un radical, soit à deux radicaux, soit aux deux extrémités d’un radical bivalent :

L’existence de deux noms est une survivance historique : en effet, les aldéhydes, comme les cétones, sont des alcools déshydrogénés, et la réaction de Piria, acétate de calcium → CaCO3 + acétone, peut engendrer des aldéhydes si l’on fait appel à un mélange des sels de calcium de l’acide R—CO2H et de l’acide formique. Si l’analogie ne s’est révélée que tardivement, c’est que les cétones ne possèdent pas le remarquable pouvoir réducteur des aldéhydes.
Nomenclature
Les aldéhydes sont désignés par le nom de l’hydrocarbure correspondant, dont l’e final fait place à la désinence al, qui porte implicitement le no 1 :

Même règle pour les cétones, mais avec la désinence one :

Mais de nombreuses autres nomenclatures existent. On peut les illustrer par deux exemples :
CH3—CHO = éthanal, aldéhyde éthylique, aldéhyde acétique, acétaldéhyde, formylméthane, méthylformaldéhyde, etc. ;
CH3—CO—CH2—CH3 = butanone, méthyl-éthyl-cétone, acétopropione, méthyl-acétone, acétyléthane, etc. Seuls les noms en italique sont pratiquement utilisés.
On peut regretter des noms vulgaires en opposition avec les règles générales de la nomenclature : l’acroléine est un aldéhyde et non une amine ; l’oxyde de mésityle est une cétone et non un éther-oxyde ; le camphre est une cétone.
État naturel
Les dérivés carbonylés sont très répandus dans le règne végétal. Beaucoup d’huiles essentielles leur doivent leur parfum ; il suffit de citer deux exemples : le principe odorant de l’essence d’amandes amères est l’aldéhyde benzoïque C6H5—CHO ; la verbénone (essence de verveine) est une cétone.
Mais la plupart des dérivés carbonylés résultent de synthèses partielles.
Préparations et modes de formation
Les dérivés carbonylés, composés au second degré d’oxydation, sont les intermédiaires entre les hydrocarbures et les alcools, d’une part, et les acides carboxyliques, d’autre part ; il est naturel de penser à des transformations réciproques utilisant soit des oxydations, soit des réductions. Mais, comme pour les autres fonctions, les méthodes proprement synthétiques, c’est-à-dire celles qui augmentent la condensation en carbone, sont les plus générales et les plus fécondes.
Ces modes de formation sont si nombreux qu’il convient de les classer et de renoncer à une énumération exhaustive.
• a) Toute fonction bivalente et plusieurs molécules au second degré d’oxydation peuvent être transformées en dérivé carbonyle. Dans le cas des dihalogénures géminés R—CX2—R′, des acétals R—C(OEt)2—R′, des dérivés azotés R—C(=Nρ)—R′, cette transformation est une simple hydrolyse. Dans le cas d’un alcyne (R—C≡CH + H2O → R—CO—CH3), il s’agit d’une hydratation ; mais le passage d’un glycol-α à un dérivé carbonylé

est une déshydratation qui peut se traduire par un réarrangement du squelette.
Souvent, ces transformations sont la phase finale d’une synthèse partant à l’origine de composés plus simples : toluène C6H5—CH3, qui est chloré en C6H5—CHCl2, hydrolysable en benzaldéhyde C6H5—CHO ; parfois, elles ne sont que la régénération d’un dérivé carbonylé transformé, par exemple, en dérivé azoté, plus facile à isoler d’un mélange.
La seule réalisation qui, dans un passé récent, présentait un intérêt pratique considérable est l’hydratation de l’acétylène en acétaldéhyde, catalysée, en particulier, par les ions H+ et Hg++.
b) Tout alcool non tertiaire peut être déshydrogéné en dérivé carbonylé :
R—CHOH—R′ → H2 + R—CO—R′.
On observe cette déshydrogénation réversible en phase gazeuse sur nickel vers 200 °C ; on peut faire appel à une oxydation ménagée (CrO3), mais, si l’alcool est primaire, il est difficile d’éviter l’oxydation de l’aldéhyde en acide.
La plupart des fonctions univalentes peuvent être oxydées au second degré : Cu(NO3)2 oxyde, en présence d’eau à 100 °C, C6H5—CH2Cl en C6H5—CHO.
c) Mais les oxydations les plus intéressantes portent sur les hydrocarbures ; le mélange H2SO4 + MnO2 oxyde le toluène en benzaldéhyde. Toutefois, dans ce domaine, les oxydations catalytiques sont de plus en plus utilisées : toluène en aldéhyde benzoïque, propène CH2=CH—CH3 en acroléine CH2=CH—CHO, méthane en formaldéhyde et surtout éthylène en acétaldéhyde (PdCl2).
d) On ne peut attendre d’une réduction la formation d’une cétone ; par contre, on pourrait espérer la réduction d’un acide R—CO2H en aldéhyde R—CHO.
En pratique, on doit faire appel à un dérivé de l’acide, par exemple le chlorure d’acide R—COCl, que l’hydrogénation catalytique, par un platine peu actif, transforme en HCl + R—CHO ; on peut aussi réduire le nitrile R—C ≡ N par le chlorure stanneux ; il en résulte l’aldimine R—CH=NH, qui s’hydrolyse en ammoniac et en aldéhyde R—CHO.
Réactions proprement synthétiques
a) Condensation d’une fonction trivalente avec un hydrocarbure, avec elle-même, avec un dérivé métallique. Nous nous bornerons à trois exemples.
1o La condensation, selon Friedel et Crafts, d’un chlorure d’acide avec un carbure aromatique :

L’inexistence du chlorure de formyle H—COCl semblerait devoir exclure l’accès à un aldéhyde ; mais le monoxyde de carbone y supplée :
2o La réaction de Piria (pyrogénation d’un sel de calcium) :

On la transforme en une réaction catalytique en envoyant les vapeurs de l’acide R—CO2H sur thorine à 350 °C :
2 R—CO2H → CO2 + H2O + R—CO—R (Senderens).
On peut faire appel à un mélange de deux acides ; si l’un est HCO2H,
R—CO2H + HCO2H → CO2 + H2O + R—CHO.
3o La condensation d’un chlorure d’acide et d’un organozincique mixte :
R—COCl + R′ZnCl → ZnCl2 + R—CO—R′ (Blaise).
On peut comparer avec l’action d’un nitrile sur un organomagnésien :

b) Alcoylation des cétones (la méthode est inapplicable aux aldéhydes, que les alcalis résinifient).
Une base très forte transforme une cétone (dont l’un des radicaux porte un hydrogène en α) en dérivé sodé ; très schématiquement
R—CO—CH2—R′ + NH2Na → NH3 + R—CO—CHNa—R′.
Un éther halohydrique R″Br forme, avec ce dérivé sodé, une cétone alcoylée :

Malheureusement, l’alcoylation n’est univoque que si les carbones qui entourent le carbonyle ne portent, en tout, qu’un seul hydrogène ; sinon on aboutit à des mélanges.




