carbonylés (dérivés) (suite)
Par contre, les dérivés carbonylés α-non saturés sont réduits par le zinc et les acides dilués, mais avec duplication :
2 CH2=CH—CHO + 2 H → CH2=CH—CHOH—CHOH—CH=CH2,
2 C6H5—CHO + 2 H → C6H5—CHOH—CHOH—C6H5.
L’hydrogénation sélective de —CO— en —CHOH— a lieu sous l’action de l’hydrure double AlH4Li.
L’hydrogénation catalytique (Ni, Pt) est régulière si le dérivé carbonyle n’est pas α-non saturé, sinon la liaison multiple s’hydrogène avant le carbonyle.
b) Addition des composés à hydrogène mobile.
1o Hydracides. Si le proton s’unit à l’oxygène,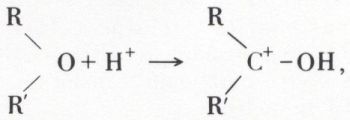
rien ne permet d’affirmer que l’ion halogénure X– s’unit au carbone.
2o Eau. L’aldéhyde formique monomère H2C=O s’unit énergiquement à l’eau :
H2C=O + H2O → H2C(OH)2 (gemglycol).
Cet hydrate constitue la majeure partie du formol (solution aqueuse de formaldéhyde). Pour les autres aldéhydes, l’hydratation, équilibrée, est de moins en moins importante au fur et à mesure que la masse molaire augmente ; elle devient négligeable pour le benzaldéhyde et l’ensemble des cétones à fonction simple ; elle est cependant totale sur le chloral :
CCl3—CHO + H2O → CCl3—CH(OH)2 (hydrate de chloral, cristallisé).
3o Alcools. Sans action sur les cétones, les alcools, tout au moins primaires, forment des hémiacétals, en présence d’une trace de HCl, puis des acétals avec les aldéhydes :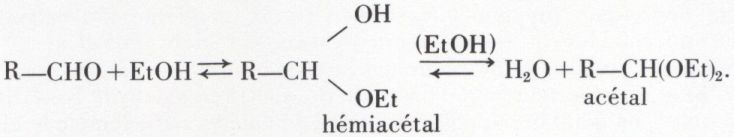
Mais les cétones sont acétalisées par les α-glycols :
4o Sulfite monosodique. Tous les aldéhydes et quelques cétones peu encombrées forment avec le sulfite monosodique des combinaisons cristallisées qui permettent de les isoler et de les régénérer par hydrolyse acide :
5o Dérivés aminogénés. L’ammoniac, les amines primaires et secondaires n’agissent qu’exceptionnellement sur les cétones. Les aldéhydes se combinent facilement à l’ammoniac et aux amines primaires :
L’imine se polymérise généralement, mais réversiblement ; toutefois, certaines imines sont stables :
C6H5—CHO + H2N—C6H5 → H2O + C6H5—CH=N—C6H5 (base de Schiff).
Par contre, si le groupe NH2 est relié à un groupe électronégatif, il agit facilement sur les aldéhydes et sur les cétones non pentasubstituées :
Si ρ = OH, on obtient l’oxime  si ρ = NH—C6H5, on obtient la phénylhydrazone
si ρ = NH—C6H5, on obtient la phénylhydrazone  etc.
etc.
Ces dérivés azotés, généralement cristallisés, identifient, par leur point de fusion, le dérivé carbonyle que leur hydrolyse régénère.
Toutes les réactions vues jusqu’ici sont facilement réversibles.
6o Composés à hydrogène mobile lié au carbone. Ce groupe comprend : l’acide cyanhydrique, les acétyléniques vrais et nombre de molécules portant un groupe —CH3, —CH2— ou =CH— relié à un accepteur mésomère (—NO2, —CHO, —CO—R, —C≡N, etc.) ; les phénols et les arylamines (hydrogènes en ortho ou en para),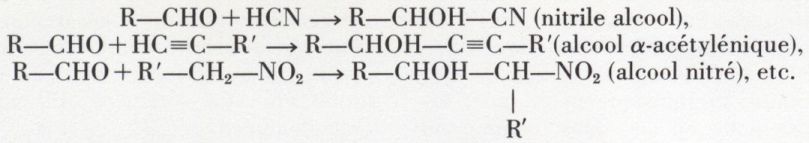
Ces condensations peuvent se répéter de deux façons.
— Si le composé antagoniste a plusieurs hydrogènes mobiles,
Ce phénol-triol subit une déshydratation intermoléculaire conduisant à une résine tridimensionnelle ; c’est le principe de la préparation des bakélites.
— Si l’alcool résultant est capable de s’anhydriser avec le réactif antagoniste,
« leucodérivé » du vert malachite, oxydable en ce colorant.
Dérivés métalliques. En principe, l’addition conduit à un alcoolate hydrolysable en alcool :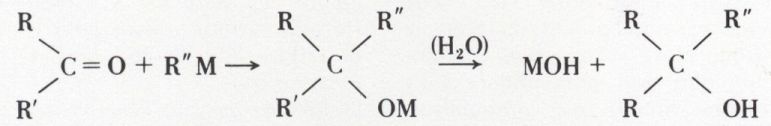
Le dérivé métallique est généralement un organomagnésien R″MgX, parfois un lithien R″Li (v. alcools).
7o Pentachlorure de phosphore. L’oxygène du carbonylé est remplacé par deux atomes de chlore géminés :
Propriétés de l’hydrogène en α
Nous appellerons dérivé carbonylé « énolisable » tout aldéhyde ou cétone portant au moins un atome d’hydrogène sur l’un des carbones liés au carbonylé ; en effet, dans ce cas, on peut envisager une prototropie :
Les énols sont rarement stables, et, le plus souvent, l’équilibre ci-dessus est si fortement déplacé vers la gauche que les critères physiques les plus sensibles ne peuvent déceler la présence de l’énol. Néanmoins, si une réaction détruit l’énol, elle déplace l’équilibre en sa faveur et le dérivé carbonylé se comporte alors comme s’il avait la formule II.
a) Cet hydrogène mobile est éliminé par les bases très fortes
[NH2Na, (CH3)3CONa].
Il en résulte un carbanion mésomère, l’ion céto-énolate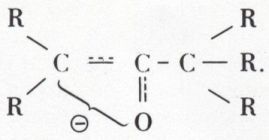
1o Cet ion attaque les éthers halohydriques R′X, et l’alcoyle se fixe au carbone, ce que l’on schématise en écrivant, comme nous l’avons fait ci-dessus, le sel de sodium
2o Il attaque les esters avec élimination d’un ion alcoyle ; par exemple,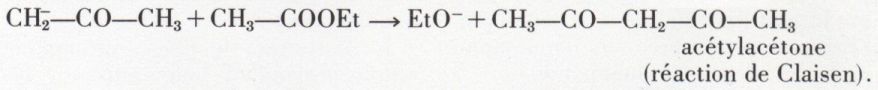
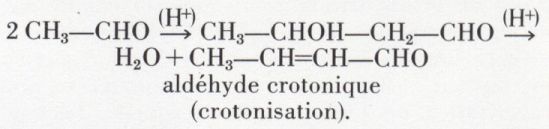
3o Il attaque le carbonylé d’une autre molécule ; par exemple,
Mais cet aldol redonne un ion énolate qui se condense avec une autre molécule d’aldéhyde, et la réaction peut se répéter ; il en résulte une condensation indéfinie qui explique la résinification des aldéhydes énolisables sous l’action des alcalis concentrés. Moins réactives, les cétones énolisables résistent mieux au milieu alcalin.
b) Cet hydrogène en α de CO est très facilement substitué par les halogènes ; s’il en existe plusieurs, ils sont le plus souvent substitués en bloc :
CH3—CHO + 3 Cl2 → 3 HCl + CCl3—CHO (chloral).
c) L’un des hydrogènes en α est facilement nitrosable :
En milieu très faiblement alcalin, on assiste à l’aldolisation (voir supra). La même réaction a lieu en milieu acide, mais l’aldol se déshydrate :