équilibre chimique (suite)
Il est cependant plus satisfaisant de disposer d’une théorie d’ensemble permettant, dans chaque cas particulier, à partir d’un petit nombre de données, une connaissance complète de l’équilibre. Cette théorie existe et constitue une belle application — faite d’abord par Josiah W. Gibbs (1875) — de la thermodynamique à la chimie. Elle conduit aux propositions fondamentales applicables à l’ensemble des équilibres et qui sont la règle des phases, la loi d’action de masses, les lois du déplacement de l’équilibre.
Règle des phases
Due à Gibbs, elle permet de déterminer a priori la variance (v. phase) des équilibres d’un ensemble donné. Elle s’écrit v = c + 2 – φ, v étant la variance, φ le nombre des phases, c celui des composants indépendants, lui-même égal au nombre des corps purs, diminué du nombre de relations, d’ordre général ou particulier, que l’existence de l’équilibre impose à l’intérieur de l’ensemble considéré. Dans le cas le moins restrictif, l’existence de l’équilibre impose une relation (la loi d’action de masses pour cet équilibre). Mais d’autres relations particulières peuvent provenir de la façon dont, par exemple, on s’impose la constitution du mélange initial ; ainsi, pour l’équilibre 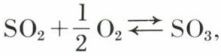 où le mélange initial est obtenu en faisant brûler du soufre dans un excès d’air, de composition O2 + 4 N2, et où les gaz sont supposés parfaits, il y a une phase, quatre corps purs présents à l’équilibre, une relation d’ordre général due à l’équilibre ; de plus, la façon dont le mélange initial de SO2, O2, N2 est constitué entraîne qu’à l’équilibre on doit avoir, quel que soit l’excès d’air initial, la relation
où le mélange initial est obtenu en faisant brûler du soufre dans un excès d’air, de composition O2 + 4 N2, et où les gaz sont supposés parfaits, il y a une phase, quatre corps purs présents à l’équilibre, une relation d’ordre général due à l’équilibre ; de plus, la façon dont le mélange initial de SO2, O2, N2 est constitué entraîne qu’à l’équilibre on doit avoir, quel que soit l’excès d’air initial, la relation 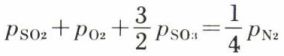 entre les pressions partielles ; cette relation, qui exprime en grandeurs d’équilibre la constante de la composition de l’air, ramène à 2 le nombre des composants indépendants ; par suite v = 3 pour l’ensemble proposé ; c’est dire que l’opérateur peut imposer la température, la pression et une des pressions partielles à l’équilibre ou, ce qui revient au même, l’excès d’air passant sur le soufre.
entre les pressions partielles ; cette relation, qui exprime en grandeurs d’équilibre la constante de la composition de l’air, ramène à 2 le nombre des composants indépendants ; par suite v = 3 pour l’ensemble proposé ; c’est dire que l’opérateur peut imposer la température, la pression et une des pressions partielles à l’équilibre ou, ce qui revient au même, l’excès d’air passant sur le soufre.
Loi d’action de masses
Elle a d’abord été établie par C. Guldberg et P. Waage (1864) pour des cas très particuliers d’équilibre monophasé (l’équilibre d’estérification-hydrolyse en est un), d’équation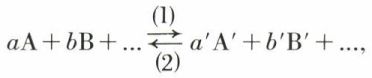
dans lesquels les réactions (1) et (2) sont simples (v. cinétique chimique) et, par conséquent, les vitesses des réactions directe et inverse sont de la forme
v1 = k1 [A]a . [B]b ..., v2 = k2 [A′]a′ . [B′]b′...,
où [A], [B]... sont les molarités, k1 et k2 les coefficients de vitesse, ceux-ci étant fonctions de la seule température. L’équilibre est réalisé lorsque v1 = v2 ; d’où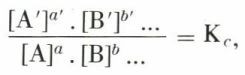
Kc = k1/k2 étant fonction de la seule température. C’est l’expression de la loi d’action de masses ; Kc est la constante d’équilibre relative aux molarités.
Cette loi très importante est démontrée en thermodynamique, dans le cas où les corps réagissants et les produits forment un système de gaz parfaits ou une solution idéale. La démonstration fait appel à la notion d’affinité chimique. Cette notion, par laquelle on a d’abord caractérisé de façon vague la tendance mutuelle des corps à entrer en réaction, a reçu, grâce à la thermodynamique, une définition mathématique précise : un système fermé qui évolue par réaction chimique entre ses constituants est, du fait de la réaction, le siège d’une transformation irréversible ; on peut, cependant, supposer que les grandeurs d’état — pression p, température T, composition des phases — restent, pour le système, déterminées à chaque instant. Le système passant d’un état à un état infiniment voisin, son entropie* éprouve une variation dS qui est la somme de deux termes ; l’un, δSe, correspond à la chaleur δQe reçue du milieu extérieur à la température T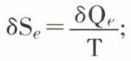
l’autre, δSir, correspond à une création d’entropie à l’intérieur du système et résultant de l’irréversibilité de l’évolution ; on a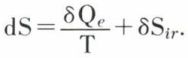
De Donder (1922) a défini l’affinité chimique 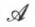 du système dans l’état considéré par l’expression
du système dans l’état considéré par l’expression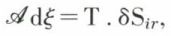
où dξ est l’accroissement, entre les deux états infiniment voisins, du degré d’avancement ξ de la réaction ; si l’équation de celle-ci est
le degré d’avancement de la réaction à l’instant donné est défini par la valeur commune des rapports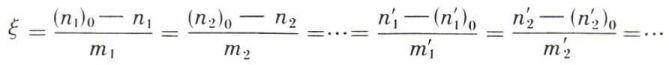
(n1)0, etc., étant les nombres initiaux de moles des corps A1 ..., et n1 ... les nombres de moles de ces mêmes corps à l’instant considéré.
Un cas pratiquement très important est celui où l’évolution est à la fois isotherme et isobare (T et p constants) : dans chacun de ses états, le système peut être caractérisé par son enthalpie* libre (potentiel* thermodynamique à pression constante, fonction de Gibbs) G = H – T . S, H étant l’enthalpie et S l’entropie. D’un état à un autre infiniment voisin, on a, si T et p sont constants, dG = dH – TdS, avec dH = δQe ; d’où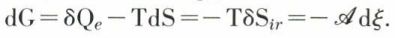
On peut donc, dans les conditions particulières imposées, écrire 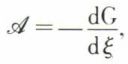 c’est-à-dire
c’est-à-dire 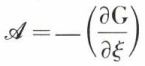 (p, T constants) ; l’affinité chimique se trouve ainsi exprimée à l’aide d’une fonction caractéristique du système, elle-même calculable de diverses façons. Une telle expression est très importante pour la prévision de l’équilibre chimique. En effet, on démontre (v. potentiels thermodynamiques) que, pour toute évolution naturelle d’un système à T et p constants — et la réaction chimique envisagée en est un exemple —, l’enthalpie libre ne peut que diminuer ; le minimum de cette fonction (ξ étant ici la seule variable) correspond donc à l’état d’équilibre ; on a alors
(p, T constants) ; l’affinité chimique se trouve ainsi exprimée à l’aide d’une fonction caractéristique du système, elle-même calculable de diverses façons. Une telle expression est très importante pour la prévision de l’équilibre chimique. En effet, on démontre (v. potentiels thermodynamiques) que, pour toute évolution naturelle d’un système à T et p constants — et la réaction chimique envisagée en est un exemple —, l’enthalpie libre ne peut que diminuer ; le minimum de cette fonction (ξ étant ici la seule variable) correspond donc à l’état d’équilibre ; on a alors 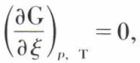 et, par suite,
et, par suite, 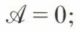 de la façon dont est définie l’affinité, l’équilibre chimique correspond à une affinité nulle.
de la façon dont est définie l’affinité, l’équilibre chimique correspond à une affinité nulle.
Pour préciser davantage l’état d’équilibre, il faut disposer d’une expression de G comme fonction, à T et p constants, du degré d’avancement de la réaction ou, ce qui revient au même, de la composition du système. Gibbs a introduit pour cela la notion de potentiel chimique. Pour un mélange homogène renfermant n1 moles du constituant A1, n2 moles de A2, etc., et qui évolue par réaction chimique, les fonctions thermodynamiques caractéristiques (énergie interne, enthalpie..., et en particulier l’enthalpie libre G) sont, à T et p constants, fonctions de n1, n2, ... ; le potentiel chimique μ1 du constituant A1 dans le mélange peut être défini comme la dérivée partielle  et il en va de même pour les autres constituants. L’enthalpie libre du mélange étant de la forme
et il en va de même pour les autres constituants. L’enthalpie libre du mélange étant de la forme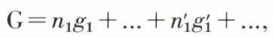
où g1 est l’enthalpie libre molaire du constituant A1 dans ce mélange, il résulte de la définition des potentiels chimiques et du théorème d’Euler que l’on a μ1 = g1, etc., T et p restant constants ; si la composition du système subit une variation infiniment petite du fait de la réaction, on a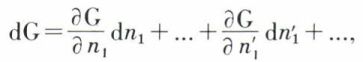
c’est-à-dire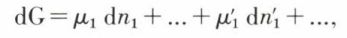
et, puisque l’on a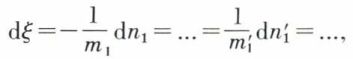
il vient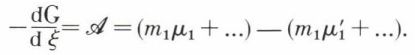
L’équilibre chimique de ce système homogène s’exprime donc par