Substances organiques renfermant deux fois le groupe carboxylique —COOH, qui sont fournies en grand nombre par la nature et la synthèse.
Introduction
Leurs préparations et leurs propriétés varient notablement en fonction de la distance des groupes carboxyliques dans la chaîne, ce qui justifie une étude séparée des différentes positions.
Il existe un seul diacide-α, l’acide oxalique HOCO—COOH ; au plus simple des diacides-β, l’acide malonique HOCO—CH2—COOH, on peut rattacher de nombreux homologues, résultant du remplacement d’un ou deux hydrogènes du groupe CH2 par un ou deux radicaux, ou bien des deux hydrogènes par les deux extrémités d’une chaîne cyclique ou par un radical bivalent. La variété des dérivés du plus simple des diacides-γ, l’acide succinique HOCO—CH2—CH2—COOH, est encore plus grande ; en effet, en dehors des éventualités envisagées pour l’acide malonique, on peut remplacer le groupe —CH2—CH2— par l’un des groupes —CH=CH ou —C=C—, ou par deux carbones voisins d’un noyau aromatique. Les diacides linéaires δ, ε, ζ..., etc., ont évidemment une parenté encore plus nombreuse.
Nomenclature
Les diacides sont désignés par le nom de l’hydrocarbure correspondant suivi du suffixe dioïque :

Mais de nombreux diacides saturés linéaires ayant des noms vulgaires :
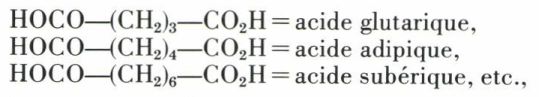
le diacide choisi comme exemple est couramment appelé acide méthyl-3 adipique.
État naturel
Beaucoup de diacides ont été rencontrés dans le règne végétal (parfois dans le règne animal), soit à l’état libre, soit à l’état de sels ou d’esters. D’autre part, des opérations simples, pyrogénation, oxydation de produits naturels, engendrent en particulier l’acide oxalique, l’acide succinique, l’acide subérique, pour ne citer que ces trois exemples.
Propriétés physiques générales
À peu près tous les diacides sont solides et fondent au-dessus de 100 °C. Les termes à moins de 10 atomes de carbone sont solubles dans l’eau bouillante et dans l’alcool, et sont généralement recristallisés dans ces solvants ; dans la série HOCO—(CH2)n–2—CO2H, les termes impairs sont solubles dans le benzène, qui dissout à peine les termes pairs.
Les diacides sont un peu plus acides que les monoacides (pKA1 ≃ 4), mais l’acide oxalique est particulier pKA1 ≃ 2, pKA2 ≃ 5. Les sels alcalins sont solubles dans l’eau, les sels de calcium insolubles au-dessus de pH = 3.
Acide oxalique HOCO—COOH
L’acide oxalique existe en traces dans la choucroute et à l’état de sel acide de potassium dans l’oseille (oxalis). Ce sel (sel d’oseille) fut la source première du diacide. L’oxalate de calcium, très insoluble, forme des concrétions (raphides) dans la racine de la bryonne et constitue certains calculs rénaux ou vésicaux. L’acide oxalique a été longtemps préparé par oxydation nitrique ou par fusion alcaline de l’amidon ou de la cellulose ; de nos jours, on fait agir l’acide sulfurique dilué sur l’oxalate de calcium de synthèse totale :
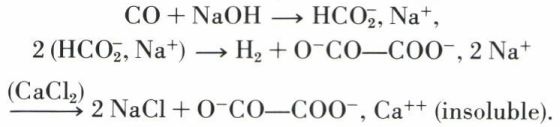
L’acide est recristallisé dans l’eau, puis dans l’alcool, qui permet d’éliminer des traces de sulfate de calcium ; on arrive ainsi à l’hydrate C2H2O4, 2 H2O, magnifiquement cristallisé et pouvant servir d’étalon alcalimétrique.
Cet hydrate perd son eau sous vide vers 100 °C. Il fond vers 103 °C en se déshydratant ; s’il est bien pur, il recristallise anhydre. L’acide anhydre fond, s’il est très pur, à 185 °C, mais, généralement, il se décompose bien avant. La décomposition est complexe ; en présence d’acide sulfurique (qui absorbe l’eau), on recueille des quantités équimolaires de CO et de CO2.
En présence de glycérine, l’acide hydraté est décarboxylé :
HOCO—COOH → CO2 + HCO2H ;
ce fut longtemps la préparation de l’acide formique.
Résistant très bien à l’acide nitrique, l’acide est oxydé par KMnO4 sulfurique en gaz carbonique, réaction stœchiométrique qui est utilisée au dosage des solutions permanganiques.
On connaît plusieurs séries de sels : sels neutres, sels acides, sels basiques ; seuls les sels alcalins sont solubles dans l’eau.
Les dérivés de la fonction acide ne sont pas tous accessibles : on ne connaît pas l’anhydride, pas plus que le monochlorure d’acide. Par contre, PCl5 conduit au chlorure d’oxalyle ClCO—COCl, qui se décompose à 100 °C :
ClCO— COCl → CO + Cl—CO—Cl (phosgène).
On connaît les monoesters et les diesters ; l’oxalate d’éthyle est transformé par PCl5 en chlorure d’éthoxalyle, lui-même instable :
ClCO— CO2Et → CO + Cl—COOEt (chloroformiate d’éthyle).
La solution aqueuse d’ammoniac transforme l’oxalate d’éthyle en oxamide NH2CO—CONH2, très peu soluble et peu fusible, que P2O5 déshydrate en cyanogène N≡C—C≡N, lequel peut, d’ailleurs, être hydrolysé en oxalate d’ammonium.
L’acétate d’éthyle se condense, en milieu alcalin, à l’oxalate d’éthyle pour former l’ester oxalacétique EtOCO—CO—CH2—CO2Et, important agent de la synthèse des acides α-cétoniques et que la chaleur décompose en ester malonique et en CO.




