




solution (suite)
Applications des lois de Raoult
Ces lois ont été à la base de méthodes efficaces de détermination expérimentale approchée de masses molaires à l’état dissous, par la mesure des effets cryométrique, ébulliométrique, et tonométrique.
La cryométrie, facile à mettre en œuvre, a été la plus utilisée, souvent à l’aide d’un appareillage rudimentaire (fig. 10), une éprouvette progressivement refroidie, mais protégée des brusques variations de température, et dans laquelle on détermine successivement, à l’aide d’un thermomètre à mercure de précision (Beckmann) gradué en centidegrés, le point de congélation du solvant pur, puis la température d’apparition des premiers cristaux du solvant dans une solution diluée, de titre connu, du corps dont la masse molaire est inconnue. La précision, bien qu’assez médiocre, est néanmoins suffisante en général pour l’identification de la substance.
Des progrès importants ont été faits en cryométrie par l’emploi d’appareils plus élaborés (fig. 11), d’un couple thermoélectrique permettant de déterminer à chaque instant l’abaissement cryométrique avec une incertitude inférieure à 10–5 °C, d’une technique différente de celle de Raoult et consistant en l’observation, dans un calorimètre adiabatique, d’un équilibre solvant solide-solution, équilibre pour lequel on mesure avec précision, par prélèvement ou par toute autre méthode, le titre actuel de la solution correspondant au Δt observé ; dans ces conditions et compte tenu de l’aspect « limite » des lois de Raoult (v. plus loin), les résultats sont précis, et la cryométrie est devenue, plus qu’un simple procédé de détermination de masses molaires, un instrument efficace et d’une portée générale pour l’étude de la structure des solutions.
L’ébulliométrie est la mesure expérimentale de l’élévation du point d’ébullition d’une solution par rapport au solvant pur sous la même pression. La pression ayant beaucoup d’influence sur les températures d’ébullition, il est avantageux que les deux déterminations soient simultanées, ce qu’on réalise à l’aide d’un ébulliomètre différentiel (fig. 12) : pour une mesure, le ballon est complètement empli de solution, de sorte que l’ébullition entraîne du liquide en même temps que de la vapeur du solvant vers l’une des soudures du couple thermoélectrique, dont la température est par conséquent celle de l’équilibre solution-vapeur du solvant ; par contre, seule la vapeur du solvant vient au contact de l’autre soudure ; se condensant en partie, elle communique à celle-ci la température d’ébullition du solvant ; le couple mesure donc la différence. On peut déduire d’une telle mesure, comme pour la cryométrie, une valeur approchée de la masse molaire du soluté.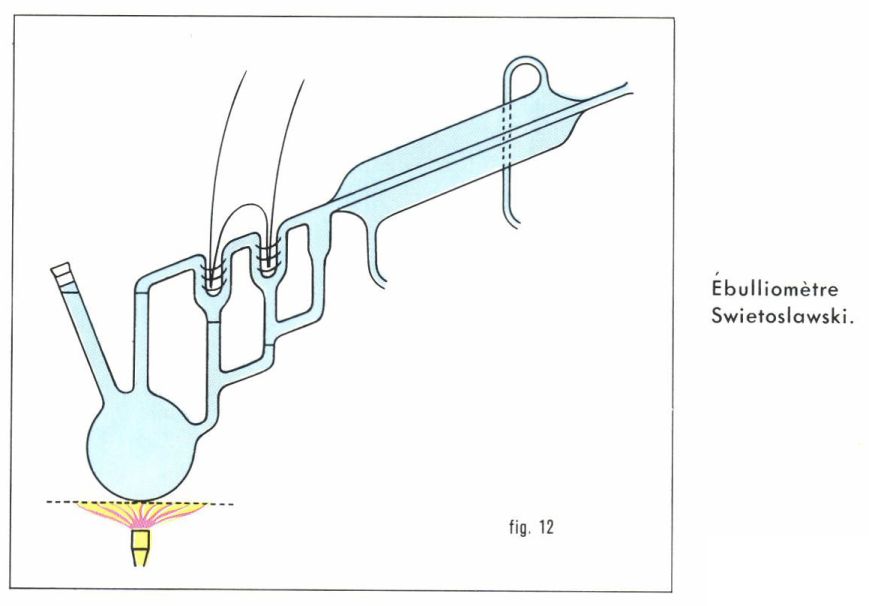
La tonométrie peut, en principe, servir à des mesures analogues ; elle n’est, cependant, guère employée, car les mesures sont délicates et peu précises.
Pression osmotique des solutions diluées, loi de Van’t Hoff
La pression osmotique fut mise en évidence par Dutrochet (1826). Un tube de verre (fig. 13), fermé en bas par une vessie de porc, ouvert en haut, contient de l’eau sucrée et plonge dans l’eau. Un équilibre s’établit par passage d’eau à travers la membrane. Quand il est réalisé, la pression hydrostatique exercée par la solution sur un côté de la membrane dépasse celle qui est exercée par l’eau de l’autre côté de la valeur π = h · ρ · g, où h est la dénivellation et ρ la masse volumique de la solution ; π est la pression osmotique de la solution. Le sucre ne traverse pas la membrane. Celle-ci est dite semi- (ou hémi-) perméable.
La pression osmotique des solutions de saccharose fut systématiquement déterminée (1877) par le botaniste W. Pfeffer, qui utilisait comme paroi semi-perméable un précipité colloïdal de ferrocyanure de cuivre, formé dans les parois d’un vase de porcelaine poreuse par la rencontre d’une solution de ferrocyanure de potassium et d’une solution de sulfate de cuivre, placées l’une à l’extérieur, l’autre à l’intérieur du vase.
Les résultats de Pfeffer furent expliqués par Van’t Hoff, qui énonça (1885) la loi de la pression osmotique : la pression osmotique d’une solution diluée non électrolysable est égale à la pression qu’exercerait le corps dissous s’il occupait, à la même température, à l’état de gaz parfait, le volume de la solution. On écrit 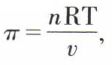 n étant le nombre de moles du corps dissous dans le volume v de solution.
n étant le nombre de moles du corps dissous dans le volume v de solution.
Comme les lois de Raoult, la loi de Van’t Hoff est utilisable, par mesure de la pression osmotique, pour calculer la masse molaire M du soluté ; en particulier, l’emploi d’osmomètres précis et le choix de la membrane permettent une bonne détermination de M pour des composés macromoléculaires, jusqu’à M = 106 environ.
On peut aisément montrer que l’existence de la pression osmotique entraîne pour une solution les effets tonométrique, ébulliométrique et cryométrique prévus par les lois de Raoult. De façon plus précise, on peut, pour en déduire les autres lois, prendre comme point de départ la loi de Van’t Hoff ou la loi tonométrique de Raoult. Il n’est donc pas étonnant que toutes ces lois présentent les mêmes caractères, dont l’un des plus importants est celui de lois limites, rigoureuses seulement pour des solutions infiniment diluées ; l’obtention de résultats précis à l’aide de ces lois oblige à effectuer des mesures pour diverses concentrations, puis à passer à la limite par extrapolation des résultats obtenus.
Une théorie thermodynamique des solutions diluées fondée sur la notion de potentiel chimique (v. équilibre chimique) permet d’établir les lois de Van’t Hoff et de Raoult. Elle suppose cependant les solutions parfaites ou idéales, analogues aux mélanges de gaz parfaits, en ce sens que les actions entre molécules du solvant A et du soluté B y sont les mêmes qu’entre molécules du solvant, ou du soluté. Dans la vapeur en équilibre avec une telle solution, la pression partielle de chaque constituant est proportionnelle à son titre molaire dans le liquide si la vapeur se comporte comme un gaz parfait :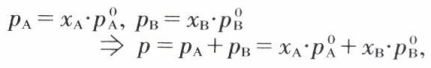
et, puisque xA + xB = 1,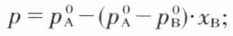
dans le diagramme isotherme (titres molaires, pression), la courbe qui représente la pression d’équilibre liquide-vapeur en fonction du titre xB du liquide est une droite ; quant à la courbe qui traduit la composition de la vapeur en équilibre avec le liquide, on a, si yB désigne le titre molaire de B dans la phase vapeur,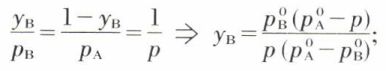
c’est un arc d’hyperbole (fig. 14) situé au-dessous de la droite, car, pour p donné, yB < xB, comme il est aisé de le vérifier dans les expressions précédentes.
En fait, bien peu de solutions peuvent être considérées comme idéales, et les écarts à l’idéalité augmentent rapidement avec le titre en soluté, ce qui oblige, dans la pratique, à ne considérer que des solutions diluées et, mieux, à passer à la limite.