Mersenne (Marin) (suite)
En ce qui concerne le domaine théorique, le P. Mersenne se livre à des spéculations — parfois un peu fastidieuses — sur les intervalles, les consonances et les dissonances, les genres, les systèmes et les modes harmoniques. Les chapitres relatifs à l’acoustique sont les meilleurs (découverte des lois des tuyaux sonores et des cordes vibrantes ; études des échos sonores ; mesure de la vitesse du son.
Mais les parties qui traitent de la pratique musicale retiennent encore l’attention des musiciens. Pour rédiger son traité de composition, son traité des instruments et son traité sur la voix et les chants, l’auteur fit appel, afin de compléter son information et de réunir les documents dont il avait besoin, à des amis musiciens : Jacques Mauduit (1557-1627) — sans doute son meilleur guide —, Jehan Titelouze* (1563-1633), Pierre Trichet (1586-1644), Jehan Basset († en 1636) [dont il publia un traité de luth], Antoine Boesset (1586-1643) et Étienne Moulinié (v. 1600 - v. 1669). Influencé par le néo-platonisme du xvie s., Mersenne croyait à l’efficacité de la musique des Anciens et à celle de la « musique mesurée ». Tout en reconnaissant que la musique italienne était plus expressive que la musique française, il restait persuadé que « la plus excellente » comporte « le sujet le plus excellent de tous, qui consiste à descrire les louanges de Dieu ».
Sa volumineuse correspondance (en cours de publication) nous renseigne, pour toutes les disciplines qui le préoccupèrent, sur sa méthode de travail.
A. V.
SOURCES. M. Mersenne, l’Harmonie universelle (Paris, 1636 ; rééd., C. N. R. S., 1964, 3 vol.). / Correspondance du R. P. Mersenne, publiée par P. Tannery et C. de Waard (P. U. F. et C. N. R. S., 1932-1970 ; 11 vol. parus).
H. de Coste, la Vie du R. P. Mersenne (S. Cramoisy, 1649). / A. Pirro, Descartes et la musique (Fischbacher, 1907). / H. Ludwig, M. Mersenne und seine Musiklehre (Halle, 1935). / R. Lenoble, Mersenne ou la Naissance du mécanisme (Vrin, 1943 ; 2e éd., 1971).
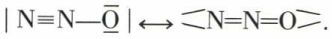
 et
et  mais aucune d’elles ne correspond exactement à toutes les caractéristiques précédentes ; l’état réel et unique de la molécule est un état mésomère, c’est-à-dire intermédiaire entre les états représentés par les formules limites précédentes, ce qu’on écrit
mais aucune d’elles ne correspond exactement à toutes les caractéristiques précédentes ; l’état réel et unique de la molécule est un état mésomère, c’est-à-dire intermédiaire entre les états représentés par les formules limites précédentes, ce qu’on écrit