Excellent portraitiste, l’effigie qu’il nous a laissée d’Agatha Van Schoonhoven (1529, Rome, galerie Doria Pamphili) démontre son sens de la mesure et l’acuité de sa vision, qui rend l’expression psychologique et le caractère social de son modèle. Van Scorel exprime le passage rapide de la Renaissance nordique vers le maniérisme italien ; initiateur dans son pays des expériences artistiques nouvelles, il eut un rayonnement considérable.
A. Z.
➙ Maniérisme.
G. J. Hoogewerff, Jan Van Scorel, peintre de la Renaissance hollandaise (La Haye, 1923) ; Jan Van Scorel en zijn navolgers en geesterverwanten (La Haye, 1941). / C. H. De Jonge, Jan Van Scorel (Amsterdam, 1940). CATALOGUE D’EXPOSITION. Werk van Jan Van Scorel (Utrecht, 1955).
Van’t Hoff (Jacobus Henricus)
Chimiste néerlandais (Rotterdam 1852 - Berlin 1911).
Celui que l’on surnommera « le Berthelot hollandais », en raison de la diversité et de l’importance de ses découvertes, s’initie à la chimie à l’université de Bonn, puis se rend à Paris, où il travaille dans le laboratoire de Charles Wurtz. Il est nommé professeur à Utrecht, enseigne ensuite à Amsterdam, puis enfin, en 1896, à Berlin, où il devient directeur de l’Institut physique de Charlottenburg. On lui doit encore la fondation de l’Institut de chimie physique d’Amsterdam, et il a été l’un des directeurs de la célèbre Revue de chimie physique, éditée à Leipzig.
Van’t Hoff est, en même temps que le Français Achille Le Bel et indépendamment de celui-ci, le créateur de la stéréochimie*. Tous deux observent en effet que seule une représentation spatiale permet de rendre compte des deux formes isomériques de l’acide tartrique, isolées en 1848 par Pasteur* ; ils montrent que les quatre liaisons de l’atome de carbone doivent occuper les sommets d’un tétraèdre régulier. Cette théorie permet de prévoir de nombreux cas d’isoméries nouvelles, que confirme l’expérience. Van’t Hoff imagine en particulier l’isomérie cis-trans des composés à double liaison pour interpréter la différence entre les acides maléique et fumarique. Il indique, en 1874, la relation existant entre les propriétés optiques et la structure des combinaisons chimiques, tous les composés présentant un carbone asymétrique devant agir sur la lumière polarisée.
Une deuxième activité de Van’t Hoff apparaît en 1884, lorsqu’il pose les fondements de la cinétique* chimique, montrant l’influence des concentrations et de la température sur les équilibres physico-chimiques. Il établit les lois quantitatives du déplacement de l’équilibre*, qu’il publie en 1885 dans son ouvrage Lois de l’équilibre, chimique.
C’est enfin en 1886 qu’il met en évidence l’extrême analogie entre les solutions diluées et les gaz parfaits. Il étudie la pression osmotique au moyen des parois semi-perméables, donne une théorie de cette pression osmotique, d’où se déduisent, conformément aux travaux de François Raoult, les diverses lois de tonométrie, d’ébulliométrie et de cryométrie.
L’ensemble de ces travaux remarquables vaut à Van’t Hoff d’être le premier titulaire du prix Nobel de chimie, en 1901.
R. T.
E. J. Cohen, Jacobus Henricus Van’t Hoff (Leipzig, 1912).
vaporisation
Passage d’un corps de l’état liquide à l’état de vapeur. (La transformation inverse est la condensation de la vapeur, ou liquéfaction*.)
La vaporisation peut avoir lieu dans le vide, ou encore en présence d’un gaz étranger. Dans le vide, la vaporisation, rapide et tumultueuse, est un phénomène interne qui présente les caractères d’une ébullition retardée ; elle est lente au contraire, n’intéressant que la surface du liquide, si celui-ci est au contact d’un gaz exerçant une pression suffisante ; c’est alors une évaporation. Dans les deux cas cependant, si le volume offert à la vapeur est limité, la vaporisation cesse quand la pression de la vapeur au-dessus du liquide atteint une valeur qui, pour un corps pur donné, ne dépend que de la température ; cette pression maximale de la vapeur est dite aussi « pression de vapeur saturante » du liquide.
Équilibre liquide-vapeur
À l’échelle microscopique, la vaporisation est le résultat de l’évasion, à travers la surface du liquide, des molécules les plus rapides, qui parviennent ainsi à échapper à l’attraction des autres molécules du liquide ; mais, à l’inverse, les molécules de vapeur qui heurtent la surface du liquide sont de nouveau absorbées par lui et se recondensent. Alors que l’évasion des molécules est un phénomène qui, pour un liquide donné, ne dépend que de la température et obéit aux lois de la thermodynamique* statistique, l’importance de la recondensation croît avec le nombre des molécules de vapeur, donc avec la pression que celle-ci exerce ; on comprend dès lors qu’un équilibre dynamique, réversible, s’établisse à température donnée, pour une certaine valeur de la pression de la vapeur.
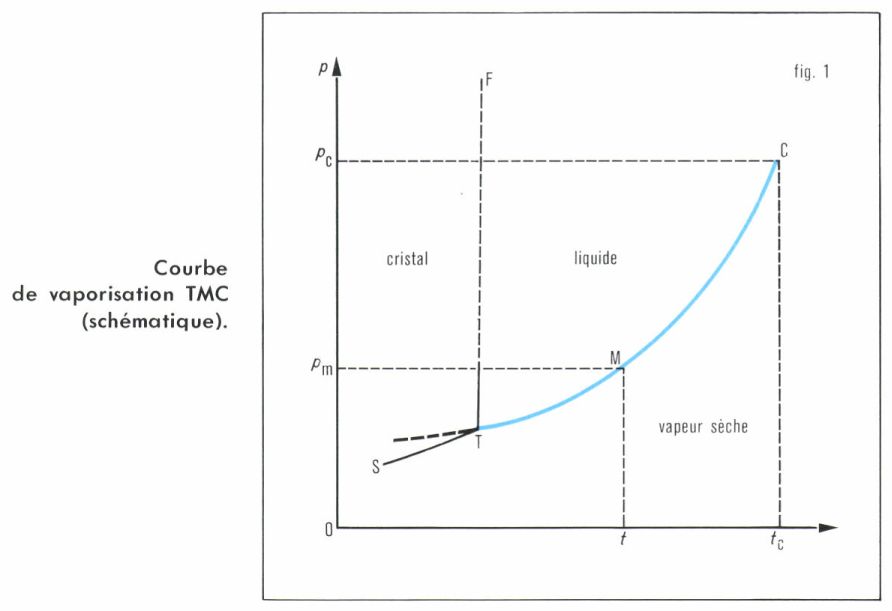
L’équilibre liquide-vapeur est univariant (1 constituant, 2 phases), ce qui entraîne que les deux facteurs de l’équilibre, pression et température, sont liés par une relation p = f (T). La représentation graphique de cette fonction constitue la courbe de vaporisation du liquide (fig. 1). Elle a pour tous les liquides les mêmes caractères essentiels, qui sont ceux de l’équilibre liquide-vapeur.
• L’ordonnée p croît avec T, en accord avec les lois du déplacement de l’équilibre, la vaporisation absorbant de la chaleur et augmentant le volume ; la croissance de p est de plus en plus rapide à mesure que la température s’élève.
La courbe est limitée : vers les températures élevées, au point critique C, où toutes les propriétés et constantes physiques deviennent les mêmes pour le liquide et sa vapeur saturante, et où s’annule la chaleur de transition ; vers les basses températures, au point triple T (v. corps pur), où la courbe de vaporisation rejoint celles de fusion et de sublimation ; toutefois, cette dernière limite n’est pas infranchissable, et l’on peut observer, à des températures inférieures à celle du point triple, des équilibres métastables entre le liquide et sa vapeur, consécutifs à une surfusion du liquide (v. fusion).